|
SCOT trial studied potential benefits over high-dose Cytoxan by Andrea Lobo, PhD | July 16, 2024  Patients with systemic sclerosis (SSc) who have a stem cell transplant see normalized gene activity related to immune function up to 4.5 years after the procedure.
That’s according to recent data from samples in the Phase 2/3 SCOT trial (NCT00114530), wherein the potential benefits of a stem cell transplant were compared with those of high-dose Cytoxan (cyclophosphamide) in severe SSc. “The normalization of the SSc [molecular] signature … parallels the clinical benefits of [stem cell transplant] at this time point and provides support for these disease-related pathways as therapeutic targets,” the researchers wrote in “Myeloablation Followed by Hematopoietic Stem Cell Transplantation and Long-Term Normalization of Systemic Sclerosis Molecular Signatures,” which was published in Arthritis & Rheumatology. SSc, or scleroderma, is an autoimmune disease where scar tissue (fibrosis) accumulates in the skin and possibly in the organs, such as the lungs, heart, or kidneys. Transplanting hematopoietic stem cells (HSC), which are precursors to all types of blood cells, has been investigated as a way to reset the immune system in people with SSc. Ferroptosis, natural block on uncontrolled cell growth, seen to be suppressed by Lindsey Shapiro, PhD | July 9, 2024  Ferroptosis, a cell death process that can help to regulate uncontrolled cell growth, was suppressed in skin cells from people with systemic sclerosis (SSc), according to recent research.
Such suppression appears to be mediated by increased activity of an antioxidant protein called GPX4. Researchers believe that targeting GPX4 to increase ferroptosis could offer a way of preventing the uncontrolled cell growth that drives scar tissue buildup, or fibrosis, in the skin of SSc patients. The study, “Upregulation of GPX4 drives ferroptosis resistance in scleroderma skin fibroblasts,” was published in Free Radical Biology and Medicine. Skin fibrosis in SSc tied to excess of fibroblasts and collagen SSc, also known as scleroderma, is an autoimmune disease wherein scar tissue accumulates in the skin and, in another disease type, in internal organs as well. Skin fibrosis is caused by the rapid and uncontrolled growth, or proliferation, of cells called fibroblasts and excessive production of collagen, a connective tissue protein. However, it is not clear what initially drives these processes, and current disease treatments aim to manage symptoms associated with scleroderma. No available therapies are specifically designed to target fibrosis in SSc, the study’s researchers stated. Programmed cell death refers to types of cell death that occur naturally and serve important biological functions. Ferroptosis is one form of programmed cell death that’s believed to help prevent the excessive cell growth that characterizes cancer. In ferroptosis, iron triggers the overproduction of toxic reactive oxygen species (ROS) molecules. This leads to a state of oxidative stress, an imbalance between ROS and the antioxidant molecules that counterbalance them, which ultimately drives cell death. As such, antioxidants can regulate ferroptosis. This form of programmed cell death has been identified as a possible therapeutic target in SSc, but the exact relationship remains to be thoroughly investigated. Updates shared at 2024 EULAR Congress may help 'better manage' SSc care by Marisa Wexler, MS | June 25, 2024  Treatment with Letairis (ambrisentan) — approved to treat pulmonary arterial hypertension (PAH), a common complication of systemic sclerosis (SSc) — may help prevent the development of PAH, or high blood pressure in the arteries of the lungs, in people with SSc.
That’s according to new data from SSc patients who completed the Phase 2 EDITA clinical trial (NCT02290613), which tested Letairis against a placebo, and who then continued their assigned regimen over a longer period. These results, along with positive data from the Phase 2 AST-MOMA trial (NCT01895244) — which evaluated a reduced-toxicity regimen of stem cell transplant in SSc patients — were presented at the 2024 European Alliance of Associations for Rheumatology (EULAR) Congress, held earlier this month in Vienna. “Taken together, these new findings could have an important bearing on the current standard of care for patients with SSc,” the alliance stated in a press release. Overall, EULAR stated that “new, strong evidence is now available to help better manage patients with this life-threatening condition,” noting, however, that “gaps remain.” Therapy is designed to block PAI-1, a mediator of inflammation, fibrosis by Patricia Inácio, PhD | June 18, 2024  The first healthy volunteers have been dosed in a Phase 1 clinical study that’s evaluating MDI-2517, a small molecule candidate to treat scleroderma and interstitial lung disease (ILD), which occurs when the lungs become scarred.
According to developers MDI Therapeutics, MDI-2517 is a potent blocker of plasminogen activator inhibitor-1 (PAI-1), a protein and key mediator of inflammation and fibrosis, or tissue scarring and thickening. “Based on comprehensive preclinical studies, this first in-human study of MDI-2517 will inform continued development of our novel, proprietary compound for the potential treatment of systemic sclerosis and interstitial lung disease, conditions where disability is significant and survival rates are poor,” Mark Weinberg, MD, chief medical officer at MDI Therapeutics, said in a company press release. In scleroderma, a self-reactive immune system causes fibrosis in the skin and possibly in internal organs, including the heart, kidney, lungs, and digestive tract. The disease is thought to develop due to a combination of genetic and environmental factors, resulting in the attack of body tissues by so-called autoantibodies. Study finds subclinical issues in 25% of patients using 'more sensitive' tool by Steve Bryson, PhD | June 11, 2024  Problems with the heart’s left ventricle at a subclinical — not yet symptomatic and overt — level were evident in 1 in every 4 people with systemic sclerosis (SSc) taking part in an Australian study when a potentially more sensitive measure than left ventricle ejection fraction was used.
Both symptomatic or asymptomatic left ventricle dysfunction also associated with poorer physical function and survival, data showed. These findings suggest “an under-appreciated burden of subtle LV [left ventricle] systolic dysfunction in SSc that has a significant impact on patient symptomatology,” the researchers wrote. The study, “Prognostic and functional importance of both overt and subclinical left ventricular systolic dysfunction in systemic sclerosis,” was published in the journal Seminars in Arthritis and Rheumatism. Left ventricle ejection fraction measures were normal in most study patients SSc, also known as scleroderma, is marked by the abnormal buildup of scar tissue (fibrosis) in the skin and, potentially, other organs like the lungs, kidneys, heart, and those of the digestive system. Heart involvement can be life-threatening in SSc, and advanced imaging techniques have shown scar tissue formation in heart muscle in up to 90% of patients without symptoms, the researchers noted. Asymptomatic myocarditis, or inflammation of the heart muscle, may also occur. Despite these abnormalities, little is known about scleroderma-related dysfunction of the left ventricle, the side of the heart that pumps oxygenated blood throughout the body. To address this gap, scientists across Australia collaborated to investigate the frequency of reduced left ventricle function in SSc. A total of 1,141 patients were recruited from the Australian Scleroderma Cohort Study, a multicenter and observational disease study. All had at least one measure taken of left ventricle ejection fraction (LVEF), the percentage of blood pumped from the left ventricle with each heartbeat. Patients with a history of secondary or non-SSc causes of left ventricle dysfunction were excluded. Nearly all participants (97.6 %) had a consistently normal LVEF of at least 50%, while 27 (2.4 %) had an LVEF of less than 50% at least once. Seven of them (0.6 %) had an LVEF of less than 40%. Those with an LVEF of less than 50% were more likely to be male, to have diffuse cutaneous SSc, a larger left atria (the top left heart chamber), higher peak blood pressure in the right ventricle, and more frequent right ventricular dysfunction. These 27 patients also were more likely to have higher levels of inflammatory markers and to more often use medicines for heart-related issues and immunosuppression. Diseases that affected skeletal muscle, those attached to bones, also were more frequent in patients with an LVEF of less than 50%. Few vaccinated patients reported symptoms worsening, according to survey by Margarida Maia, PhD | June 4, 2024  More than half of people with systemic sclerosis (SSc) who hesitated to get vaccinated against COVID-19 were concerned they might have a flare-up, but very few vaccinated patients have reported their symptoms worsening, according to a 2021–2022 survey study.
“Even though the peak of the pandemic has passed, COVID-19 infections and hospitalizations continue to present substantial risk for vulnerable people with autoimmune rheumatic diseases, including SSc,” said the study’s researchers, who noted that patients who are unsure about a vaccination should be informed of its safety. The study, “COVID-19 vaccinations and infections among individuals with systemic sclerosis: a Scleroderma Patient-centered Intervention Network (SPIN) Cohort study,” was published in Seminars in Arthritis and Rheumatism. When vaccines against COVID-19 were made available, there was little information on their safety in people with autoimmune rheumatic diseases. Concerns that getting one may have serious side effects led many people to hesitate. Treatment is ‘promising option,’ more studies needed, researchers say by Margarida Maia, PhD | May 14, 2024 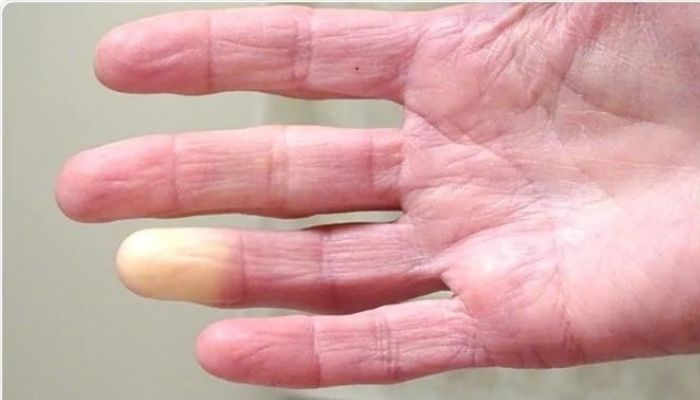 Local injection with botulinum toxin type A (BTX-A), such as Botox, may ease the severity of pain associated with Raynaud’s phenomenon in people with scleroderma, also known as systemic sclerosis, a meta-analysis study found.
The study, “Efficacy and safety of botulinum toxin in treating scleroderma-associated raynaud’s phenomenon: a systematic review and meta-analysis,” was published as a research letter in the Archives of Dermatological Research. Scleroderma affects the connective tissue that holds tissues and organs together. While it mainly causes the skin to harden, scleroderma also can damage internal organs and blood vessels. It often starts with Raynaud’s phenomenon, which may appear long before other symptoms. Raynaud’s phenomenon limits blood flow to fingers and toes due to cold or stress. As a result, fingers and toes can change color and become numb or prickly. This is often accompanied by pain and sometimes digital ulcers or sores. Bacteria-derived BTX-A, often injected locally to smooth wrinkles on the face, among other uses, is being tested as a treatment for Raynaud’s phenomenon secondary to scleroderma. It’s thought that it may work in part by widening the blood vessels and reducing pain. Formula could ID patients in need of 'enhanced managing strategies,' study says by Lindsey Shapiro, PhD | April 30, 2024  Researchers have developed a model for predicting which systemic sclerosis (SSc) patients with interstitial lung disease (ILD) — together known as SSc-ILD — will experience progressive lung function declines that are known to be associated with a poorer prognosis.
The prediction algorithm accounts for nine clinical factors, including age, certain SSc features and symptoms, therapy use, and disease-associated antibodies. All were found to be linked to the development of this aggressive form of ILD — called progressive fibrosing interstitial lung disease or PF-ILD — in a group of about 300 patients. “This study developed the first prediction model for PF-ILD in patients with SSc-ILD,” the researchers wrote, noting that “the established formula … could help in identifying patients with the highest risk, who should receive enhanced managing strategies during follow-up.” Titled “Prediction of progressive fibrosing interstitial lung disease in patients with systemic sclerosis: insight from the CRDC cohort study,” the work was published in RMD Open. Half of patients in study found to have aggressive disease type PF-ILD In SSc, also called systemic scleroderma, an overactive immune system drives inflammation and scar tissue buildup — known as fibrosis — in the skin and internal organs. When the lungs are affected, patients may experience ILD, then named SSc-ILD, where the lung tissue is scarred and damaged, and breathing becomes more difficult. The severity and progression of interstitial lung disease can vary. PF-ILD, a progressive form, is marked by “rapidly aggravating [shortness of breath], progressive deteriorating lung function, declining physical functional capacity, worsening health-related quality of life, adverse therapeutic response and often early mortality,” the researchers wrote. An earlier identification of which patients are likely to experience this type of progression may enable a sooner start to interventions that could slow it down or stop it, preventing irreversible lung damage, according to the team. While some studies have reported a number of potential predictive factors of PF-ILD, an accurate model for identifying SSc patients who might be at a high risk of the aggressive lung disease has yet to be developed. Now, a team of scientists in China sought to develop one such model by looking at clinical information from 304 SSc-ILD patients who were registered in a Chinese database. Meta-analysis has implications for prognosis and prevention, researchers sayby Andrea Lobo, PhD | April 23, 2024  Having scleroderma increases the risk of developing diseases affecting blood vessels of the brain or heart, known as cerebrovascular and cardiovascular diseases, a review study found.
Relative to controls, the risk of stroke — caused by poor blood flow to the brain — in scleroderma patients was 64% higher, whereas that of cardiovascular disease was 112% higher. “Our findings support increased clinical surveillance and consideration of preventative strategies in this high-risk population,” researchers in Taiwan wrote. Their study, “Association between systemic sclerosis and risk of cerebrovascular and cardiovascular disease: a meta-analysis,” was published in Scientific Reports. Scleroderma, also known as systemic sclerosis (SSc), is characterized by the excessive production of collagen that leads to the accumulation of scar tissue, affecting the skin and potentially internal organs, such as the digestive tract and lungs. The disease may also damage blood vessels, both small and large, leading to complications such as pulmonary arterial hypertension and scleroderma renal crisis. Evidence has suggested that people with scleroderma may have an increased risk of stroke, heart attack, and peripheral vascular disease, which is reduced circulation in blood vessels outside the brain and heart. However, “the risk of cerebrovascular and cardiovascular complications attributed to SSc remains debated,” the researchers wrote. Calcitonin and SOST levels may point to PAH and ILD, small study find by Marisa Wexler, MS | April 9, 2024  Levels of two blood proteins, calcitonin and SOST, may be markers of lung disease in people with scleroderma, a new study shows.
“This study indicates that serum calcitonin and SOST levels are promising biomarkers for [scleroderma]-related PAH [pulmonary arterial hypertension] and ILD [interstitial lung disease], respectively,” the researchers wrote, though they noted that “further research is needed to verify these results and understand the underlying mechanisms.” The study, “Protein profiling in systemic sclerosis patients with different pulmonary complications using proteomic antibody microarray,” was published in Arthritis Research & Therapy. Scleroderma, also called systemic sclerosis or SSc, is marked by abnormal scarring that usually affects skin and can also affect various other organs throughout the body. When the disorder involves the lungs, SSc can lead to complications like interstitial lung disease (ILD, marked by lung scarring) and pulmonary arterial hypertension — PAH, defined by high pressure in the blood vessels that carry blood to and through the lungs. These lung issues are leading causes of mortality in SSc, and early detection is key to facilitate optimal treatment. |
Scleroderma Association of Queensland
©Scleroderma Association of Queensland. All rights reserved. Website by Grey and Grey.